Research
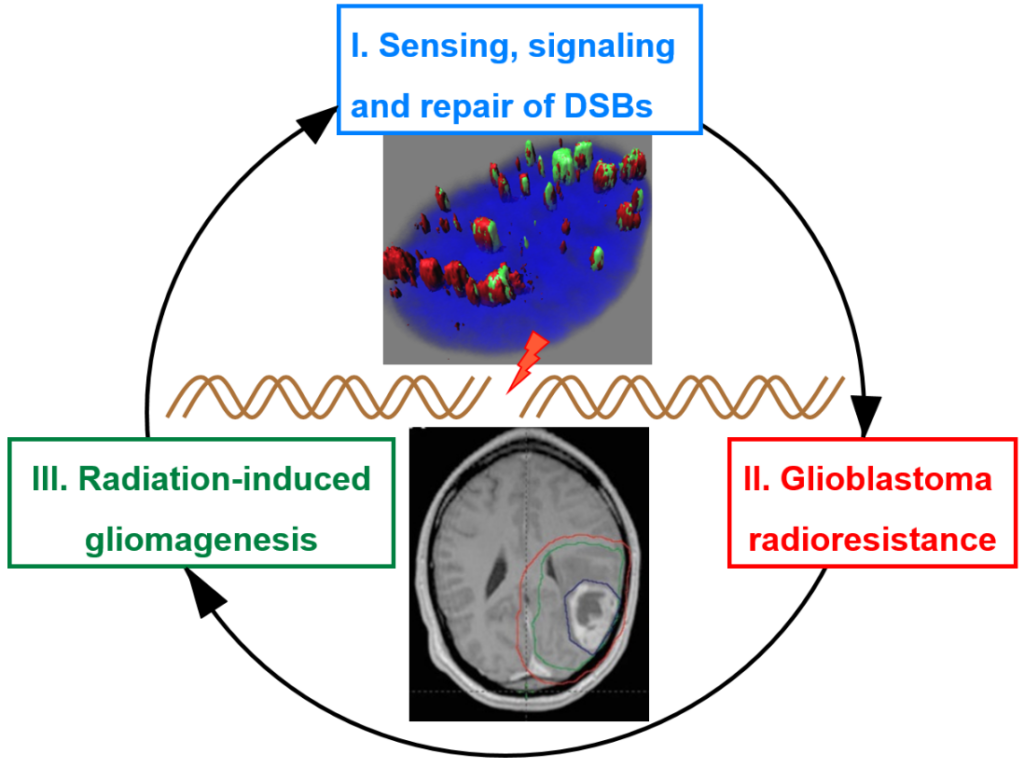
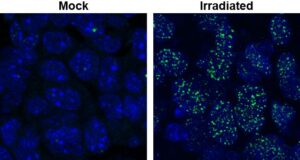
1. DNA double-strand break sensing, signaling, and repair. We are interested in cellular events that occur in response to DNA breaks, with an emphasis on DNA end resection. Our early research re-defined our understanding of the DDR with regard to the exact roles of the three kinases that are activated by these DSBs – ATM, ATR, and DNA-PKcs. Our research uncovered the existence of an “ATM-independent” pathway of ATR activation and cell cycle checkpoint implementation involving DSB resection by a 5’-3’ exonuclease, EXO1. Subsequently, we established that DNA end resection by EXO1 is a critical step which influences the choice that a cell makes between DNA repair pathways (NHEJ vs HR) and between cell cycle checkpoint activation modes (ATM vs ATR). Importantly, we established that EXO1 was phosphorylated and regulated by CDKs 1/2 in S/G2 phases of the cell cycle. This work is significant as it demonstrates how HR is linked to the cell division cycle. Subsequently, we established that DNA-damage induced degradation of EXO1 is a mechanism by which the cell negatively regulates DNA end resection. This work is significant because it demonstrates how negative regulation of resection is necessary for optimal HR.
More recently, in collaboration with Patrick Sung’s laboratory at UT Health, we solved the conundrum of why eukaryotic cells possess multiple resection nucleases by showing that different nucleases have differing specificities for DSBs based on the nature of other DNA lesions adjacent to the break. Also, in collaboration with the Weixing Zhao’s laboratory at UT Health, we established that the ubiquitin ligase activity of BRCA1-BARD1 is critical for DNA end resection and subsequent steps in HR. Regulatory mechanisms impinging on EXO1 or other resection enzymes are of significance from a carcinogenesis standpoint as multiple SNPs in these nucleases have been linked to a host of human cancers. Moreover, disrupting accurate repair pathway choice can be an effective cancer therapeutic strategy as shown by us in the context of gliomas (see below).
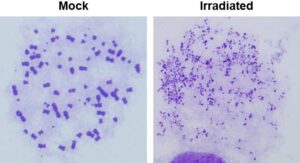
2. Glioblastoma radioresistance. An important translational goal of our research is to understand the genetic basis of glioblastoma (GBM) radio- and chemo-resistance with the objective of developing rational therapeutic-sensitization strategies for these lethal brain tumors. We first systematically examined the contributions of key GBM-specific genetic alterations to therapy resistance using state-of-the-art transgenic mouse models, with a focus on two key events driving gliomagenesis – amplification of EGFRvIII and loss of PTEN.
This research helped formulate an important concept in radiation oncology, i.e., key genetic changes occurring during carcinogenesis impact DNA repair pathways in specific ways. Specifically, EGFRvIII amplification and activation of the PI3K-Akt pathway augments NHEJ via DNA-PKcs while PTEN loss results in attenuated HR. Importantly, we discovered that the dual PI3K/mTOR inhibitor NVP-BEZ235 blocks both ATM and DNA-PKcs activation, thereby attenuating both NHEJ and HR pathways of DSB repair in glioma cells. Using pre-clinical mouse models, we showed that the drug can cross the blood-brain-barrier, and block DSB repair in gliomas thus sensitizing these tumors to IR and improving survival of tumor-bearing mice. Subsequently, we established that GBMs being treated with the chemotherapeutic agent TMZ acquire chemoresistance, in part, due to augmented HR, and that such tumors can be re-sensitized using CDK inhibitors (based upon results from our EXO1 project). Our research established that GBMs (or other cancers) may be vulnerable to tailored therapies with specific DNA repair inhibitors based upon their DNA repair status. Our work has resulted in the development of several novel approaches that can be used to augment GBM therapy, illustrating how our work on basic mechanisms of DNA repair can synergize with our translational research.
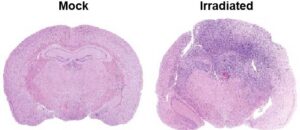
3. Radiation-induced glioblastoma development. Complementing our research on IR as a therapeutic agent, a third project in our laboratory focuses on the carcinogenic effects of radiation, especially particle radiation. We are studying cellular responses to complex DNA damage inflicted by the HZE (High-Z, High-Energy) particles and their gliomagenic consequences. With funding from NASA, we developed two complementary mouse models of radiation-induced glioblastoma (RIG), essentially compound transgenic mice with brain restricted tumor suppressor deletions, that do not develop tumors spontaneously but are susceptible to transformation after radiation exposure. Our research with these mice established that low doses of HZE particles are highly transforming while gamma or X-rays are also very gliomagenic but only at higher doses. We discovered that loss of the Ink4a-Rb and Arf-p53 tumor suppressor axes and amplification of the MET proto-oncogene are critical events in the transformation process. We also found a linear correlation between atomic number of charged particles and GBM development in these mouse models.
These findings are very pertinent to NASA as the decision to launch future long-duration space missions hinges upon the accurate estimation of cancer risks to astronauts from these particles. Moreover, this research is also very relevant to cancer therapy due to the increasing use of charged particles (protons and carbon) for targeted tumor therapy in the clinic. This project has led to the development of valuable mouse models that could be used to understand mechanisms underlying radiation induced GBM development and recurrence and to test radiation countermeasures.
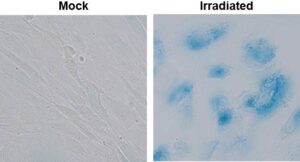
4. Therapy-Induced Senescence (TIS) and Glioblastoma Recurrence. Cellular senescence is primarily an anti-cancer barrier, but, paradoxically, can also have tumor promoting roles. Recent research shows that senescence-associated secretory phenotype (SASP) factors secreted by senescent cells can promote inflammation and tumor development (antagonistic pleiotropism). In fact, senescence is now classified as an “emerging hallmark” of cancer.
We are working on mechanisms underlying radiotherapy-induced senescence in the brain tumor microenvironment and the deleterious consequences thereof. We find that TIS of normal brain cells and even tumor cells can drive glioblastoma recurrence via secretion of SASP factors. We are currently dissecting mechanisms underlying the crosstalk between senescent and nonsenescent cells in brain tumors, and devising strategies to blunt GBM recurrence using senolytic therapy.